CP2K: Hybrid Gaussian and Plane-Wave DFT for Large-Scale Materials Simulations
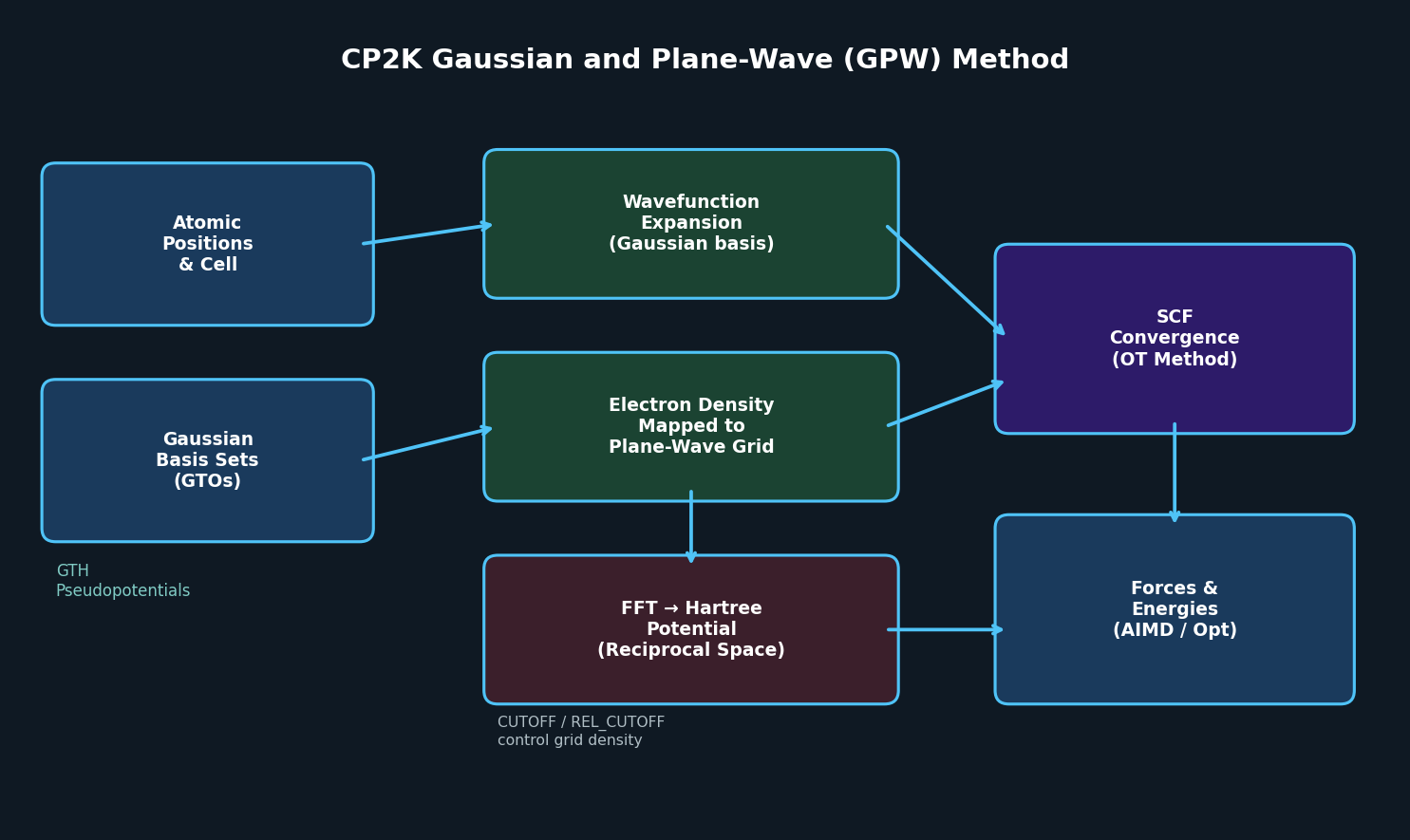
CP2K has established itself as one of the most capable open-source codes for large-scale quantum chemistry and condensed-matter simulations. Unlike conventional plane-wave DFT codes such as VASP or Quantum ESPRESSO, CP2K employs the Gaussian and Plane-Wave (GPW) method — a dual-basis approach that combines the compactness of Gaussian-type orbitals (GTOs) with the efficiency of an auxiliary plane-wave basis for the electron density. This hybrid strategy enables linear-scaling DFT calculations on systems containing thousands of atoms, making CP2K uniquely suited for problems at the interface of molecular and materials science.
The GPW Method: Why It Matters
Traditional plane-wave DFT codes represent both wavefunctions and electron density on a uniform real-space grid. This is accurate but computationally expensive for large, low-density systems such as liquids, amorphous materials, surfaces with adsorbates, and biological interfaces. CP2K's GPW approach separates these concerns:
- Wavefunctions are expanded in localized Gaussian basis sets (DZVP, TZV2P, etc.), which are compact and chemically intuitive.
- Electron density is mapped onto an auxiliary plane-wave grid for efficient computation of the Hartree potential via fast Fourier transforms (FFT).
- Pseudopotentials (Goedecker-Teter-Hutter, GTH) are optimized for use with Gaussian bases, ensuring smooth, accurate core representations.
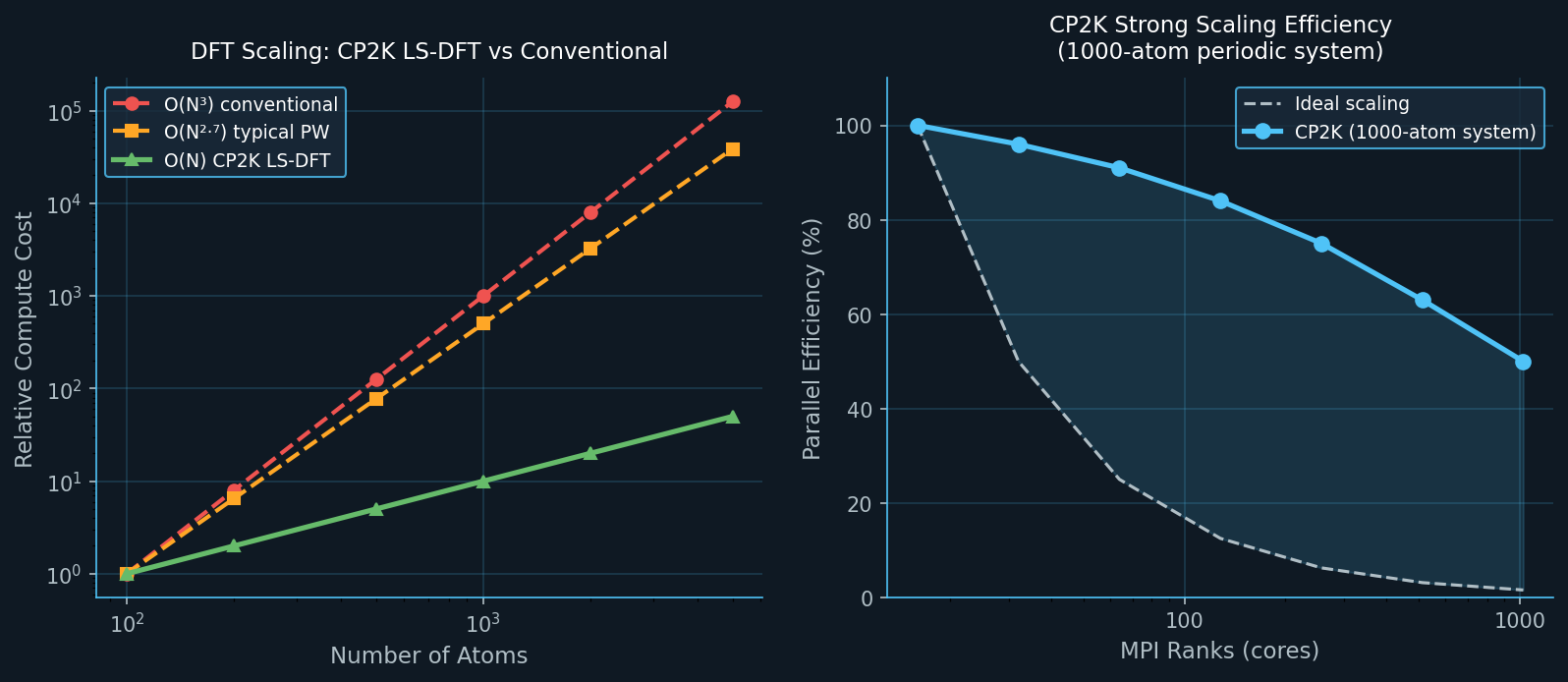
The result is a code that scales as O(N) for large systems using the Linear Scaling (LS-DFT) module, while retaining the accuracy of conventional DFT for smaller systems. For metallic systems or cases requiring exact exchange, CP2K also supports the GAPW (Gaussian Augmented Plane-Wave) method, which adds all-electron accuracy for core states.

Key Capabilities for Materials Scientists
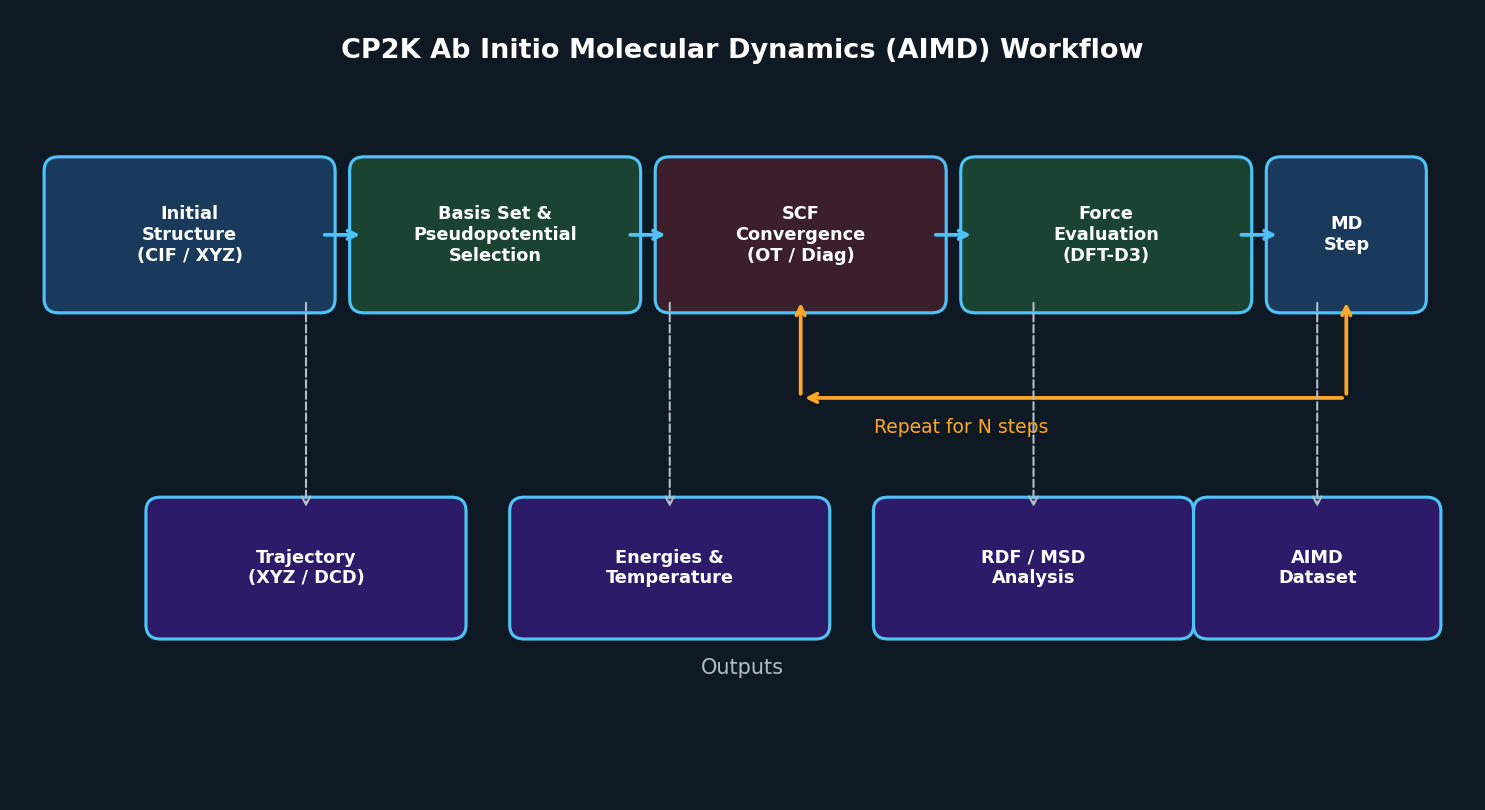
Ab Initio Molecular Dynamics (AIMD)
CP2K's most widely used feature is its Born-Oppenheimer and Car-Parrinello molecular dynamics engines. AIMD in CP2K is production-ready for:
- Liquid-state simulations: Water, ionic liquids, molten salts, and electrolyte solutions at DFT accuracy.
- Solid-liquid interfaces: Electrode-electrolyte interfaces for battery and fuel-cell research.
- Amorphous materials: Glass formation, disorder in oxides, and amorphous silicon.
The code supports a rich ensemble of thermostats (Nosé-Hoover chains, CSVR, GLE) and barostats (MTK, Parrinello-Rahman) for NVT and NPT dynamics.
Hybrid Functionals and Range-Separated Exchange
CP2K implements the Auxiliary Density Matrix Method (ADMM) to make hybrid functional calculations (PBE0, HSE06) tractable for large periodic systems. ADMM approximates the exact exchange contribution using a smaller auxiliary basis, reducing the computational cost by an order of magnitude compared to conventional hybrid DFT. This makes band-gap-corrected calculations feasible for systems with hundreds of atoms — a regime where VASP or CASTEP hybrid calculations become prohibitively expensive.
Linear-Scaling DFT for Thousands of Atoms
The LS-DFT module in CP2K uses the density matrix-based approach with orbital transformation (OT) methods. Key features include:
- Sparse matrix algebra exploiting the locality of Gaussian basis functions.
- Purification algorithms (McWeeny, TC2) for idempotent density matrices.
- Efficient parallelization across thousands of MPI ranks with optional OpenMP threading.
Systems of 1,000–10,000 atoms are routinely studied, including nanoparticles, grain boundaries, and large biomolecule-surface complexes.
Machine Learning Potentials Integration
CP2K integrates with DeePMD-kit and supports the NequIP and MACE neural network potential frameworks through its MOTION/MD interface. This allows seamless active-learning workflows: run short AIMD trajectories in CP2K to generate reference data, train an MLP, then validate against new CP2K single-point calculations — all within a single scripted pipeline.
Practical Workflow: Setting Up a CP2K AIMD Simulation
A typical CP2K input file (*.inp) is organized into sections. Below is a representative structure for an NVT AIMD run of a liquid system:
&GLOBAL
PROJECT water_nvt
RUN_TYPE MD
PRINT_LEVEL LOW
&END GLOBAL
&FORCE_EVAL
METHOD Quickstep
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
&MGRID
CUTOFF 400
REL_CUTOFF 50
&END MGRID
&QS
EPS_DEFAULT 1.0E-12
&END QS
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-6
MAX_SCF 50
&OT ON
MINIMIZER DIIS
&END OT
&END SCF
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&VDW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
TYPE DFTD3
PARAMETER_FILE_NAME dftd3.dat
REFERENCE_FUNCTIONAL PBE
&END PAIR_POTENTIAL
&END VDW_POTENTIAL
&END XC
&END DFT
&END FORCE_EVAL
&MOTION
&MD
ENSEMBLE NVT
STEPS 5000
TIMESTEP 0.5
TEMPERATURE 300
&THERMOSTAT
TYPE CSVR
&CSVR
TIMECON 100
&END CSVR
&END THERMOSTAT
&END MD
&END MOTIONKey parameters to tune:
- CUTOFF: The plane-wave cutoff for the auxiliary grid (300–600 Ry for most systems).
- REL_CUTOFF: Controls the mapping of Gaussians to the grid (40–60 Ry is typical).
- TIMESTEP: 0.5 fs for systems with light atoms (H); up to 2 fs for heavier systems.
Performance and Parallelization
CP2K scales efficiently on modern HPC clusters. The GPW method's FFT operations parallelize well across distributed memory, while the OT SCF solver benefits from dense linear algebra (ScaLAPACK/ELPA). For AIMD production runs:
- Small systems (< 200 atoms): 16–64 cores, wall times of minutes per MD step.
- Medium systems (200–1,000 atoms): 128–512 cores, ~1–5 minutes per step.
- Large LS-DFT (1,000–5,000 atoms): 512–4,096 cores with near-linear scaling.
The ELPA eigensolver library is strongly recommended over ScaLAPACK for systems above ~500 atoms, providing 2–4× speedups in the diagonalization step.
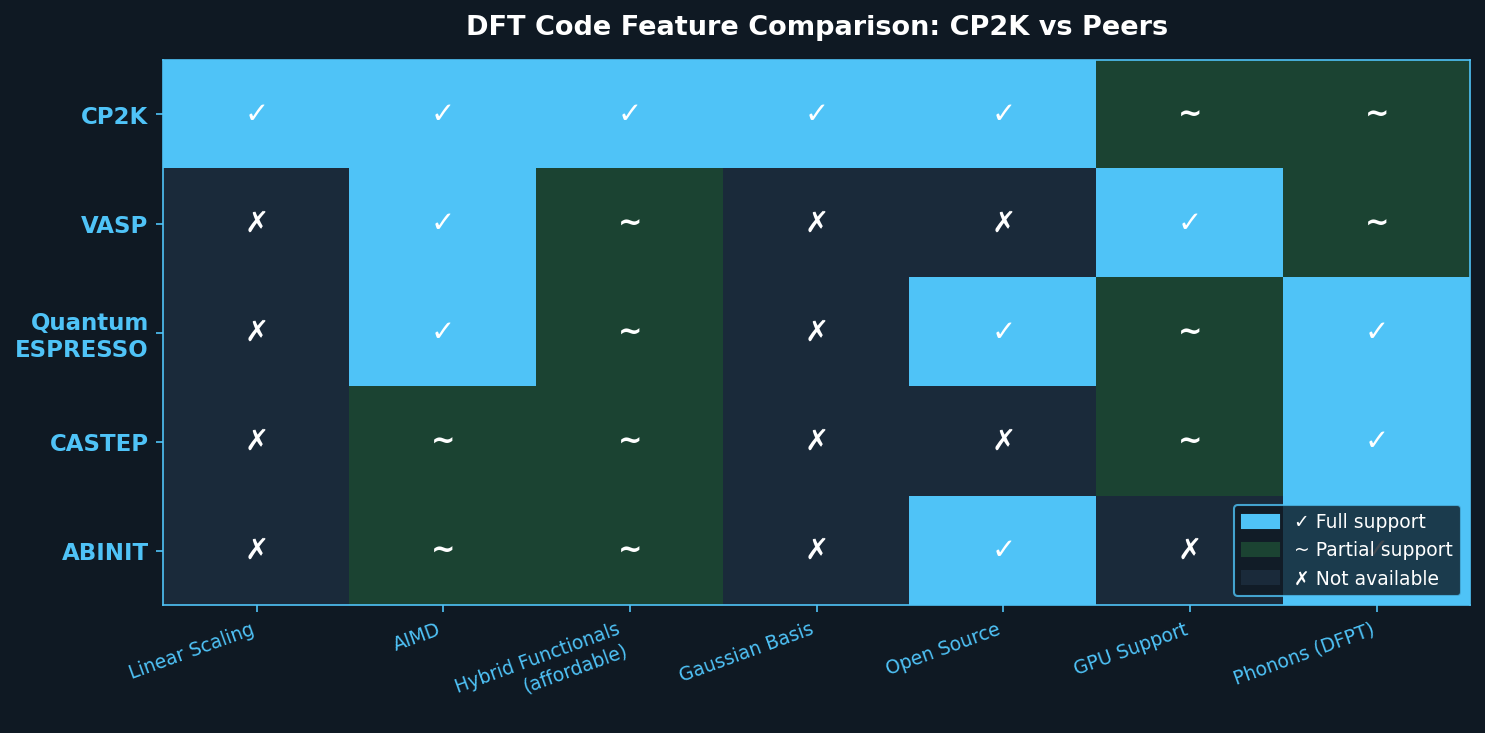
Comparison with Peer Codes
| Feature | CP2K | VASP | Quantum ESPRESSO |
|---|---|---|---|
| Basis type | Gaussian + PW | Plane-wave | Plane-wave |
| Linear scaling | Yes (LS-DFT) | No | No |
| AIMD | Yes (BOMD, CPMD) | Yes (BOMD) | Yes (BOMD) |
| Hybrid functionals | Yes (ADMM) | Yes | Yes (limited) |
| Cost (license) | Free/open-source | Commercial | Free/open-source |
| Best for | Large, low-density systems | Periodic solids | Periodic solids |
Getting Started
- Official website: https://www.cp2k.org
- Input reference: https://manual.cp2k.org
- Basis sets and pseudopotentials: Distributed with the CP2K source in
data/ - Community forum: https://groups.google.com/g/cp2k
- Tutorials: https://www.cp2k.org/howto
CP2K's combination of linear-scaling DFT, production-quality AIMD, and affordable hybrid functionals makes it an indispensable tool for materials scientists working on large, chemically complex systems. Its open-source nature and active community ensure continued development aligned with emerging HPC architectures, including GPU acceleration via the DBCSR sparse tensor library and CUDA/HIP backends.