
GROMACS for Materials Science: Advanced Molecular Dynamics Beyond Biomolecules

While GROMACS (GROningen MAchine for Chemical Simulations) is widely recognized for biomolecular simulations, its powerful molecular dynamics engine has increasingly found applications in materials science. This versatile open-source package offers exceptional performance for simulating polymers, liquid crystals, nanocomposites, and interfacial phenomena—domains where atomic-scale insights drive innovation.
Why GROMACS for Materials?
GROMACS excels in materials applications through several key capabilities. Its highly optimized algorithms leverage modern CPU and GPU architectures, enabling simulations of systems containing millions of atoms. The code's efficient neighbor searching and domain decomposition make it particularly suitable for large-scale polymer systems and heterogeneous materials where long-range interactions matter.
The software supports a wide range of force fields beyond biomolecular ones, including OPLS-AA, TraPPE, and custom parameterizations for synthetic polymers, ionic liquids, and inorganic materials. This flexibility allows researchers to model everything from polyethylene crystallization to graphene-polymer nanocomposites with appropriate interaction potentials.

Advanced Analysis Tools for Materials Properties
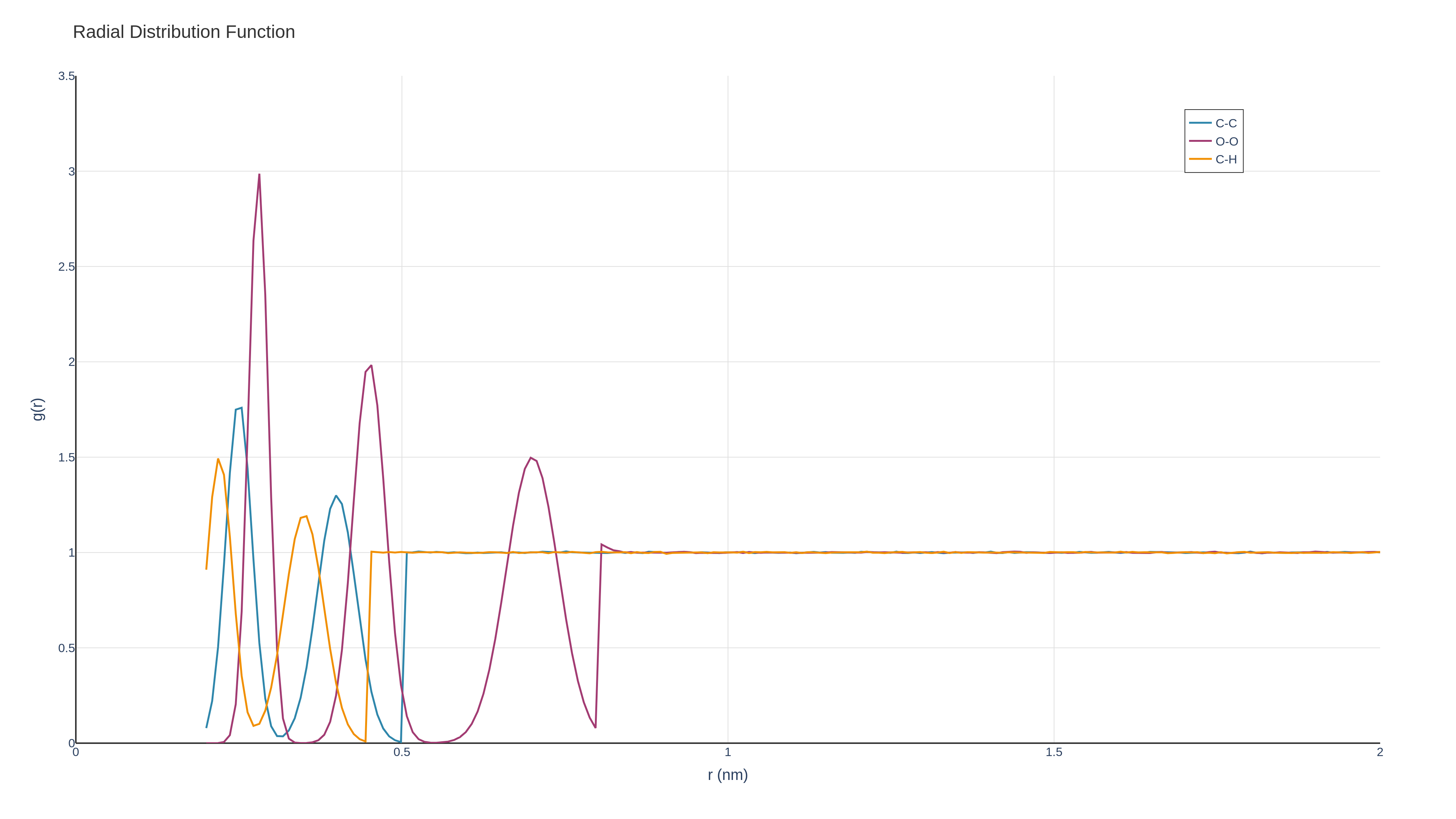
GROMACS provides sophisticated analysis utilities that extend well into materials characterization. The gmx rdf command computes radial distribution functions essential for understanding local structure in amorphous materials and liquids. For polymer scientists, gmx polystat calculates end-to-end distances, radius of gyration, and persistence lengths—critical metrics for characterizing chain conformations.
The gmx msd tool enables calculation of mean-square displacements, directly yielding diffusion coefficients through the Einstein relation. This proves invaluable when studying ion transport in solid electrolytes or polymer membranes for battery and fuel cell applications. Additionally, gmx energy extracts thermodynamic properties like heat capacity and thermal expansion coefficients from simulation trajectories.
Free Energy Calculations and Phase Behavior
One of GROMACS's most powerful features for materials science is its implementation of free energy perturbation and thermodynamic integration methods. These techniques enable precise calculation of solvation free energies, partition coefficients, and chemical potentials—quantities that govern phase equilibria and miscibility in polymer blends and composites.
The umbrella sampling module, combined with the weighted histogram analysis method (WHAM), allows researchers to map free energy landscapes for processes like polymer crystallization, nanoparticle aggregation, or molecular adsorption on surfaces. Such calculations provide mechanistic insights that complement experimental phase diagrams and guide materials design.
Interfacial Phenomena and Surface Chemistry
GROMACS handles interfacial systems with particular elegance. Its support for anisotropic pressure coupling enables simulation of surfaces, thin films, and membranes under realistic mechanical conditions. Researchers studying polymer-metal interfaces, liquid-vapor equilibria, or nanoparticle surface functionalization benefit from tools like gmx density for computing density profiles perpendicular to interfaces.
The software's constraint algorithms (LINCS, SHAKE) maintain molecular geometry while allowing efficient integration timesteps, crucial when simulating stiff bonds in materials like carbon nanotubes or ceramic surfaces. Combined with virtual sites for handling rigid bodies, GROMACS can model complex hierarchical materials from atomistic to mesoscale regimes.

Integration with Materials Workflows
Modern materials research demands integration across multiple simulation scales and tools. GROMACS interfaces seamlessly with quantum chemistry packages like CP2K and ORCA for QM/MM simulations, enabling studies of catalytic sites or defects in materials. Its trajectory formats are compatible with visualization tools like VMD and OVITO, facilitating publication-quality rendering of simulation results.
Python bindings through MDAnalysis and PyMOL enable custom analysis workflows, while the PLUMED plugin adds enhanced sampling methods like metadynamics—essential for exploring rare events in materials transformations. This ecosystem makes GROMACS a central hub in computational materials science pipelines.
Performance Optimization for Large Systems
For materials scientists working with large systems, GROMACS offers unparalleled performance tuning options. The gmx tune_pme utility automatically optimizes the particle-mesh Ewald parameters for electrostatic calculations, often yielding 20-30% speedups. GPU acceleration through CUDA and OpenCL enables routine simulations of 100+ nanosecond trajectories for systems with hundreds of thousands of atoms.
The recent implementation of GPU-resident algorithms keeps data on accelerators throughout the simulation, minimizing CPU-GPU transfer overhead. This architectural advance makes GROMACS competitive with specialized materials codes while maintaining its flexibility and extensive feature set.
Best Practices and Resources
Success with GROMACS in materials applications requires attention to force field validation and equilibration protocols. The GROMACS manual (https://manual.gromacs.org) provides comprehensive documentation, while the user mailing list offers community support. For materials-specific guidance, the GROMACS tutorials repository includes examples for polymer systems and surface simulations.
Researchers should benchmark their specific systems, as optimal simulation parameters vary with material type and properties of interest. Starting with well-validated test cases from the literature ensures reliable results before tackling novel materials challenges.
GROMACS continues to evolve as a premier tool for materials simulation, bridging the gap between specialized biomolecular codes and materials-focused packages. Its combination of performance, flexibility, and robust analysis capabilities makes it an essential component of the modern computational materials scientist's toolkit.
Further Reading:
- GROMACS Official Documentation: https://www.gromacs.org
- GROMACS Tutorials: https://tutorials.gromacs.org
- Materials-specific force fields: https://virtualchemistry.org