LAMMPS: Large-Scale Molecular Dynamics Simulation for Materials Science
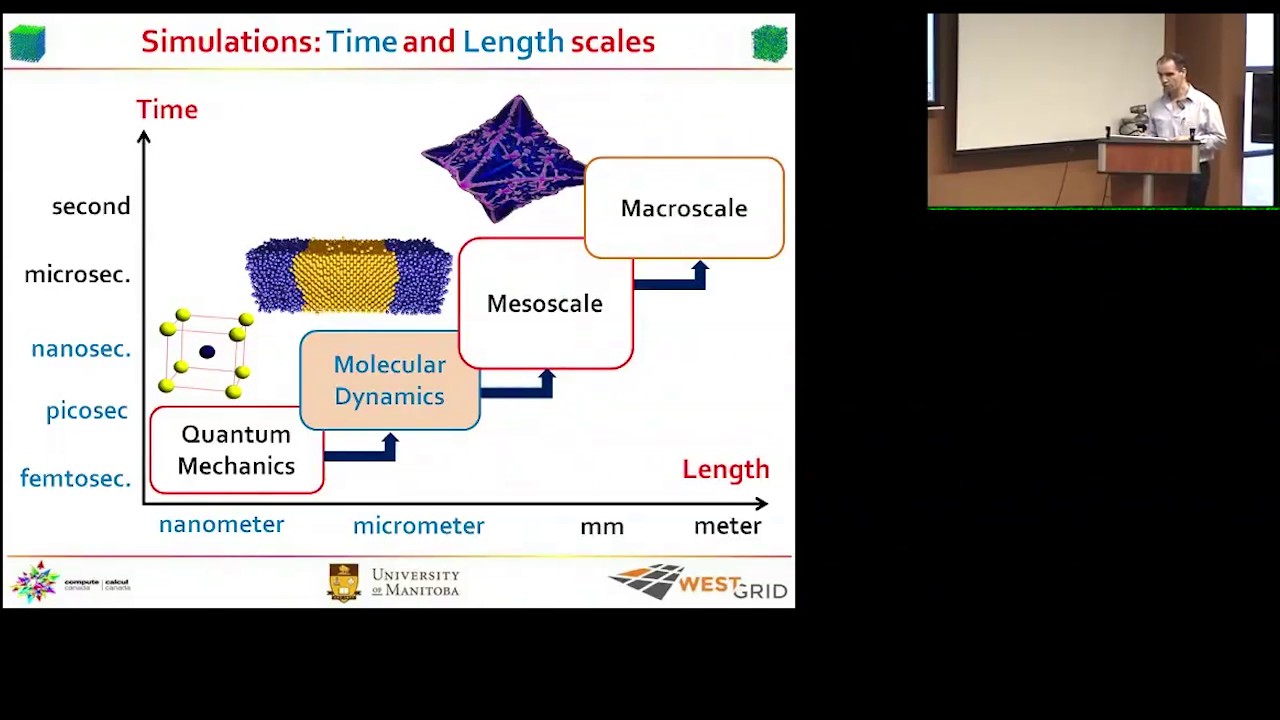
LAMMPS, an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator, stands as a preeminent classical molecular dynamics (MD) code, meticulously developed by Sandia National Laboratories. Since its release as an open-source code in 2004, LAMMPS has evolved into an indispensable tool for particle-based modeling across an expansive spectrum of materials science and computational chemistry applications.
Overview and Philosophy
LAMMPS is designed for the simulation of particle ensembles in various states—liquid, solid, or gaseous—and is adept at handling atomic, polymeric, biological, metallic, granular, and coarse-grained systems through a diverse array of force fields and boundary conditions. The core philosophy behind LAMMPS is its focus on materials modeling, enabling the simulation of systems ranging from the atomic to the mesoscale and even continuum levels.
The software is distinguished by its remarkable flexibility, versatility, scalability, and robust performance, all underpinned by its open-source nature and a vibrant, supportive community. LAMMPS is freely available under the terms of the GNU General Public License (GPLv2) and boasts high portability, running seamlessly on Linux, Windows, and macOS platforms.

Architecture and Performance
LAMMPS leverages the Message Passing Interface (MPI) for parallel communication, facilitating high-performance simulations on single processors or massively parallel computing environments, often employing spatial decomposition of the simulation domain. Furthermore, it supports accelerated performance on various hardware, including CPUs, GPUs, and Intel Xeon Phis, enabling researchers to leverage modern computing hardware for faster and larger simulations.
Comprehensive Features and Capabilities
LAMMPS is a classical molecular dynamics code endowed with a rich and diverse set of functionalities, broadly categorized into general features, particle and model types, interatomic potentials, atom creation, ensembles, constraints, boundary conditions, integrators, diagnostics, output, and specialized features.
General Features
LAMMPS is engineered for high-performance computing, capable of executing simulations on a single processor or in parallel. Its parallelization capabilities are multifaceted, encompassing distributed memory message-passing parallelism via MPI and shared memory multi-threading parallelism through OpenMP. It employs spatial decomposition of the simulation domain for MPI parallelism and particle decomposition within spatial decomposition for OpenMP and GPU parallelism.
As an open-source project, LAMMPS is distributed under the GPLv2 license, fostering a collaborative development environment. The codebase is highly portable, written in C++-11, with optional packages potentially requiring C++17. Its modular design means that most functionalities are encapsulated within optional packages, allowing users to tailor the build to their specific needs.
Particle and Model Types
LAMMPS offers extensive flexibility in defining the types of particles and models within a simulation, controlled primarily via the atom_style command. It supports a wide array of particle representations, including:
- Atomic systems: Basic atoms for simple materials
- Coarse-grained particles: Such as bead-spring polymers for soft matter
- United-atom polymers or organic molecules: Where groups of atoms are represented as single interaction sites
- All-atom polymers, organic molecules, proteins, DNA: Detailed representations including every atom
- Materials-specific models: For metals, metal oxides, and granular materials
- Mesoscopic models: Coarse-grained mesoscale models
- Finite-size particles: Including spherical and ellipsoidal particles, line segments (2D), triangles (3D), and rounded polygons (2D) and polyhedra (3D)
- Specialized particles: Point dipole particles and particles with magnetic spin
- Rigid bodies: Collections of particles treated as a single rigid entity
- Hybrid combinations: Allowing for the simultaneous use of different particle and model types within a single simulation
Interatomic Potentials (Force Fields)
A cornerstone of any molecular dynamics simulation is the interatomic potential, or force field, which describes the interactions between particles. LAMMPS provides an exceptionally broad range of potentials:
Pairwise Potentials: Standard potentials including Lennard-Jones, Buckingham, Morse, Born-Mayer-Huggins, Yukawa, soft, Class II (COMPASS), hydrogen bond, harmonic, gaussian, tabulated, and user-scripted potentials, as well as Coulombic and point-dipole interactions.
Many-body Potentials: These potentials account for interactions involving more than two particles simultaneously, crucial for accurate modeling of metals and semiconductors. Examples include EAM (Embedded Atom Method), Finnis/Sinclair, MEAM (Modified Embedded Atom Method), Stillinger-Weber, Tersoff, REBO (Reactive Empirical Bond Order), AIREBO, ReaxFF, and COMB.
Machine Learning Potentials (MLPs): LAMMPS is at the forefront of integrating MLPs, offering built-in support for ACE, AGNI, GAP, Behler-Parrinello (N2P2), POD, RANN, and SNAP. Furthermore, it provides interfaces to ML potentials distributed by external groups, such as ANI, CHIMES, DeepPot, HIPNN, and MTP.
Long-range Interactions: For systems with charges, point-dipoles, and Lennard-Jones dispersion, LAMMPS implements various methods including Ewald summation, Wolf summation, PPPM (Particle-Particle Particle-Mesh), MSM, and ScaFaCoS.
Specialized Potentials: Including coarse-grained potentials for mesoscopic systems (DPD, Gay-Berne, REsquared, colloidal, DLVO, oxDNA/oxRNA, SPICA), water potentials (TIP3P, TIP4P, SPC, SPC/E), and interlayer potentials for graphene and analogous 2D materials.
Atom Creation and System Setup
LAMMPS provides flexible methods for setting up initial atomic configurations:
- Reading from files: Atom coordinates can be read directly from existing data files
- Lattice generation: Atoms can be created on predefined lattices, useful for constructing crystalline structures
- Manipulation: Geometric or logical groups of atoms can be deleted or displaced
- Replication: Existing atoms can be replicated multiple times to expand the system size
- External tools: For complex molecular designs, external software like Avogadro for molecular drawing and Packmol for arranging molecules are commonly used
Ensembles, Constraints, and Boundary Conditions
LAMMPS offers comprehensive control over simulation conditions:
- Dimensionality: Supports 2D or 3D systems
- Simulation Domain: Orthogonal or non-orthogonal (triclinic symmetry) simulation domains
- Thermodynamic Ensembles: Integrators for constant NVE, NVT, NPT, and NPH ensembles
- Thermostatting and Pressure Control: Various options for controlling temperature and pressure
- Constraints: Including harmonic constraint forces, rigid body constraints, SHAKE/RATTLE for bond and angle constraints
- Monte Carlo Methods: Supporting bond breaking, formation, swapping, and template-based reaction modeling
- Boundary Conditions: Various types including advanced Open Boundary Condition (OBC) methods
Output and Analysis Capabilities
LAMMPS offers robust output capabilities:
- Log File: Records thermodynamic information such as temperature, pressure, volume, and energy
- Dump Files: Store atom coordinates, velocities, and other per-atom quantities
- Restart Files: Binary files for seamless continuation of simulations
- Parallel I/O: Supports parallel I/O for dump and restart files
- Per-atom Quantities: Can output energy, stress, centro-symmetry parameter, Common Neighbor Analysis (CNA), etc.
- Analysis Features: Custom partitioning for binning, spatial and time averaging, and histogramming capabilities
- Snapshot Formats: Various formats including native, XYZ, XTC, DCD, CFG, NetCDF, HDF5, ADIOS2, and YAML
Applications Across Scientific Domains
LAMMPS's versatility makes it applicable across a broad range of scientific and engineering disciplines, particularly in materials science and computational chemistry.
Materials Science
LAMMPS is extensively utilized in materials science to investigate the properties of various materials:
- Mechanical properties: Elasticity, plasticity, fracture, and deformation mechanisms
- Phase transitions: Simulating melting, solidification, and other phase changes
- Defect dynamics: Studying the behavior of dislocations, vacancies, and grain boundaries
- Nanomaterials: Designing and optimizing carbon nanotubes, graphene, and nanocomposites
- Complex systems: Modeling solid-state materials, soft matter, and coarse-grained systems
Biological Systems
In computational chemistry and biophysics, LAMMPS is a powerful tool for modeling biomolecular systems:
- Proteins: Protein folding, dynamics, and interactions
- DNA: DNA structure and dynamics
- Lipid bilayers: Membrane dynamics and interactions
- Large biomolecular systems: Designed for high-performance simulation
Chemical Engineering
LAMMPS finds applications in chemical engineering for:
- Chemical reactions: Modeling reaction mechanisms at the atomic level
- Diffusion processes: Studying mass transport and diffusion in various media
- Complex fluids: Understanding the behavior of polymers, colloids, and other complex fluids
- Material development: Supporting the development of new materials and processes
Physics Applications
In physics, LAMMPS is employed to study fundamental phenomena:
- Heat conduction: Investigating thermal transport properties
- Shock waves: Simulating material response under extreme conditions
- Phase behavior: Analyzing phase transitions and critical phenomena
- Quantum-Mechanical insights: Bridging quantum mechanics and continuum mechanics through multi-scale modeling
Best Practices for LAMMPS Simulations
Effective utilization of LAMMPS for molecular dynamics simulations involves several key steps and considerations, from molecular design to high-performance computing.
System Preparation and Design
Molecular Design: The initial step involves creating all necessary molecules and ions using suitable software. Avogadro software can be used to draw molecular structures, which can then be energy-minimized to achieve their most stable state.
System Arrangement: After designing individual molecules, they must be arranged within a simulation box. The Packmol package is commonly used for this purpose, requiring an input script to define the number of molecules, ions, and their desired locations.
Visualization: Before running the simulation, it is crucial to visualize the initial system configuration to verify its correctness. Tools like OVITO (Open Visualization Tool) or VMD (Visual Molecular Dynamics) are indispensable for this step.
Simulation Execution and Configuration
Unit System Selection: Choosing the correct unit system (e.g., lj, real, metal, si) is crucial for accurate simulations.
Force Field Selection: Specifying the appropriate interatomic potentials (pair, bond, angle, dihedral, improper styles) based on the system being studied.
Thermodynamic Conditions: Defining temperature, pressure, and ensemble (NVE, NVT, NPT) appropriate for the research objectives.
Simulation Parameters: Including timestep, run duration, and output frequency settings.
High-Performance Computing Optimization
For systems with a large number of atoms, running simulations on personal computers is often impractical due to time constraints. LAMMPS is designed for efficient execution on HPC resources:
- Single Processor:
lmp -in input_script.in - Multi-Processor (Parallel Computing):
mpirun -np x lmp -in input_script.in, where x is the number of CPU cores
Performance Optimization Strategies
GPU Acceleration: Leveraging CUDA, OpenCL, HIP, or SYCL for compatible potentials and features.
Parallel Computing: Efficient use of MPI and OpenMP, including static and dynamic load-balancing.
Memory Optimization: Managing memory usage, especially for large systems.
Optional Packages: Building LAMMPS with only necessary optional packages to reduce compilation time and executable size.
Community Support and Resources
LAMMPS benefits from an active user community and comprehensive documentation:
- Documentation: The official LAMMPS documentation provides detailed explanations of features, commands, installation guides, tutorials, and examples
- User Support: Forums, Slack channels, and IRC channels are available for user support
- Tutorials: Comprehensive tutorials covering basics and advanced techniques
- Workshops: Regular workshops and symposia for community engagement
Advanced Features and Specialized Capabilities
Multi-replica Models
LAMMPS includes advanced capabilities for multi-replica simulations, essential for exploring complex energy landscapes:
- Nudged elastic band (NEB)
- Hyperdynamics
- Parallel replica dynamics
- Temperature accelerated dynamics
- Parallel tempering
- Path-integral MD (PIMD)
Specialized Applications
Load-balancing: Static and dynamic load-balancing with optional recursive bisectioning decomposition.
Aspherical Particles: Generalized aspherical particles for complex geometries.
Stochastic Rotation Dynamics (SRD): For mesoscale fluid simulations.
Visualization: Real-time visualization and interactive MD capabilities.
QM/MM Coupling: Hybrid Quantum Mechanics/Molecular Mechanics coupling for multi-scale modeling.
Peridynamics: Modeling of peridynamics for fracture mechanics.
Conclusion
LAMMPS stands as a comprehensive and powerful molecular dynamics simulation platform that has revolutionized computational materials science and related fields. Its open-source nature, extensive feature set, and active community support make it an invaluable resource for researchers across diverse scientific domains.
The platform's ability to handle systems ranging from simple atomic structures to complex biomolecular assemblies, combined with its scalability from desktop computers to supercomputing clusters, positions it as a cornerstone technology for modern computational research. Its continuous development and integration of cutting-edge techniques, including machine learning potentials and advanced simulation methods, ensure its continued relevance in addressing the evolving challenges of materials science and nanotechnology.
By adhering to best practices in system preparation, simulation execution, and performance optimization, researchers can effectively leverage LAMMPS to gain atomic-level insights into material behavior, driving innovation in materials design, drug discovery, and fundamental scientific understanding.