
Materials Studio CASTEP: Plane-Wave DFT for Crystalline Materials
Materials Studio CASTEP represents one of the most robust implementations of plane-wave density functional theory (DFT) specifically optimized for crystalline materials simulation. Developed by the Castep Developers Group and commercialized through Dassault Systèmes BIOVIA, this simulation engine has become an industry standard for predicting structural, electronic, and optical properties of periodic systems with exceptional accuracy.
Core Capabilities and Architecture
CASTEP employs pseudopotentials and plane-wave basis sets to solve the Kohn-Sham equations, making it particularly well-suited for systems with translational symmetry. Unlike localized basis set approaches, plane-wave methods ensure systematic convergence and eliminate basis set superposition errors—critical advantages when computing formation energies, phonon spectra, or elastic constants.
The code supports ultrasoft pseudopotentials and norm-conserving pseudopotentials, with on-the-fly pseudopotential generation capabilities. This flexibility allows researchers to balance computational efficiency against accuracy requirements for specific elements and properties. For heavy elements, CASTEP implements relativistic treatments including scalar relativistic and spin-orbit coupling corrections.

Advanced Property Calculations
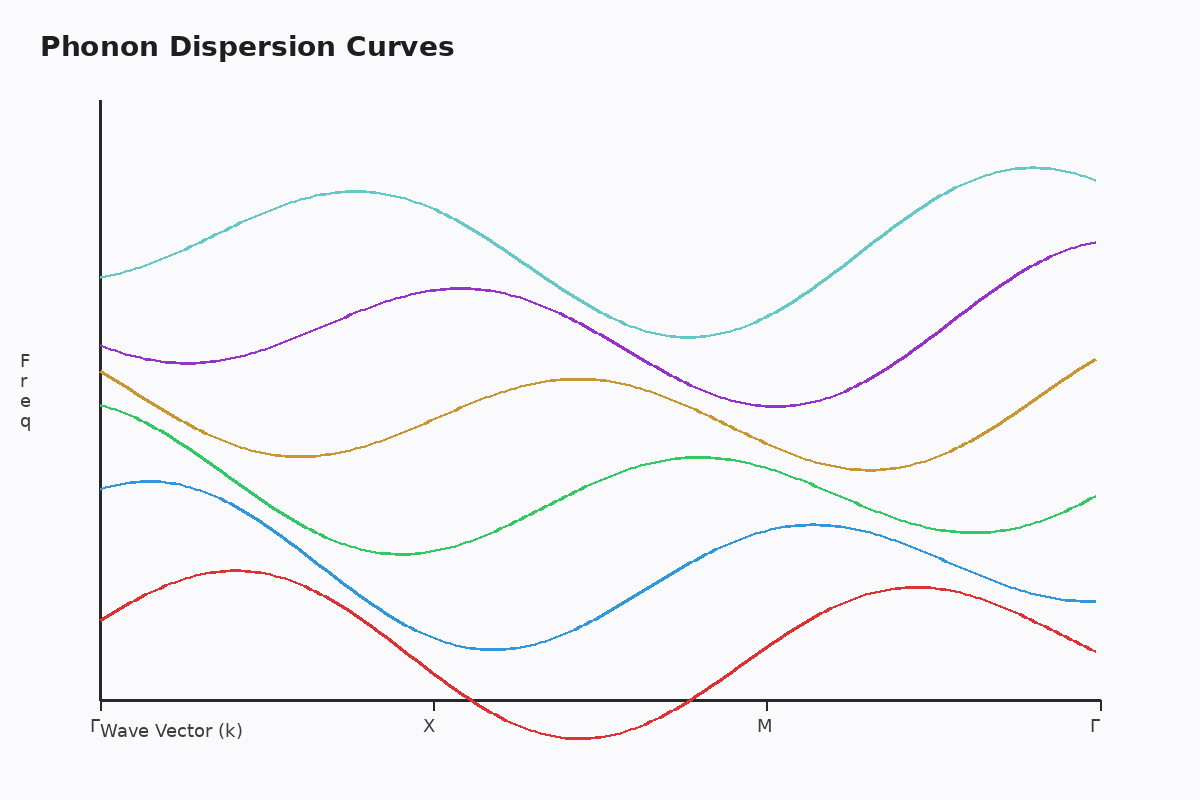
One of CASTEP's distinguishing features is its comprehensive suite of property prediction modules. Beyond standard ground-state calculations, the software excels at computing:
Phonon Dispersion and Thermodynamics: Using density functional perturbation theory (DFPT), CASTEP calculates phonon frequencies without requiring finite-displacement supercells. This approach provides access to thermodynamic properties including heat capacity, entropy, and free energy as functions of temperature—essential for phase stability analysis.
Optical and Spectroscopic Properties: The code computes dielectric functions, absorption spectra, and refractive indices through time-dependent DFT or the random phase approximation. For experimentalists, CASTEP can simulate NMR chemical shifts, EPR g-tensors, and core-level spectroscopy, enabling direct comparison with characterization data.
Mechanical Properties: Elastic constant tensors, bulk moduli, and stress-strain relationships emerge naturally from CASTEP's stress tensor implementation. The geometry optimization algorithms handle both internal coordinates and cell parameters simultaneously, facilitating structure prediction under pressure.
Practical Workflow Considerations
CASTEP integrates seamlessly with the Materials Studio graphical interface, lowering the barrier to entry for researchers less familiar with command-line workflows. However, the underlying engine runs efficiently on high-performance computing clusters through its MPI parallelization scheme, scaling effectively to hundreds of cores for large unit cells.
Convergence testing remains critical: users must systematically verify that energy cutoffs (typically 300-800 eV depending on pseudopotentials) and k-point sampling densities yield converged results for their properties of interest. The Monkhorst-Pack scheme generates k-point grids, with denser meshes required for metals and narrow-gap semiconductors.
For geometry optimization, CASTEP offers multiple algorithms including BFGS and damped molecular dynamics. The choice depends on the system—BFGS converges rapidly for well-behaved structures, while damped dynamics proves more robust for complex energy landscapes with multiple local minima.
Integration with Materials Discovery Workflows
CASTEP's strength lies in its integration with broader materials informatics pipelines. The software interfaces with crystal structure databases, enabling high-throughput screening of candidate materials. Researchers have successfully employed CASTEP in machine learning workflows, using DFT-computed properties as training data for predictive models.
The code's Python scripting interface (through Materials Studio or standalone) facilitates automation of repetitive tasks—structure generation, batch calculations, and post-processing. This programmability proves invaluable when exploring compositional spaces or optimizing dopant configurations.
Limitations and Complementary Tools
While CASTEP excels for crystalline systems, its plane-wave approach becomes inefficient for isolated molecules or systems with large vacuum regions. In such cases, localized basis set codes like Gaussian or NWChem may prove more appropriate. Additionally, standard DFT functionals (LDA, GGA) suffer from well-known limitations for strongly correlated systems and van der Waals interactions—though CASTEP does implement dispersion corrections and hybrid functionals to partially address these issues.
For researchers requiring explicit many-body treatments or excited-state dynamics beyond linear response, complementary tools like VASP with GW capabilities or real-time TDDFT codes become necessary.
Conclusion
CASTEP remains a cornerstone tool for computational materials scientists working with crystalline systems. Its combination of rigorous plane-wave methodology, comprehensive property prediction capabilities, and accessible interfaces makes it particularly valuable for bridging computational predictions with experimental validation. As materials design increasingly relies on predictive simulation, CASTEP's role in accelerating discovery—from battery electrodes to photovoltaic absorbers—continues to expand.
For detailed documentation and tutorials, consult the official CASTEP documentation and the Materials Studio user guides. The CASTEP academic consortium also maintains extensive resources for researchers at educational institutions.