
Quantum ESPRESSO for First-Principles Materials Simulation: Density Functional Theory in Practice
Quantum ESPRESSO (opEn-Source Package for Research in Electronic Structure, Simulation, and Optimization) has emerged as one of the most widely adopted open-source codes for first-principles electronic structure calculations in materials science. Built on density functional theory (DFT) and plane-wave pseudopotential methods, this powerful suite enables researchers to predict material properties from fundamental quantum mechanics without empirical parameters.
Core Capabilities and Architecture
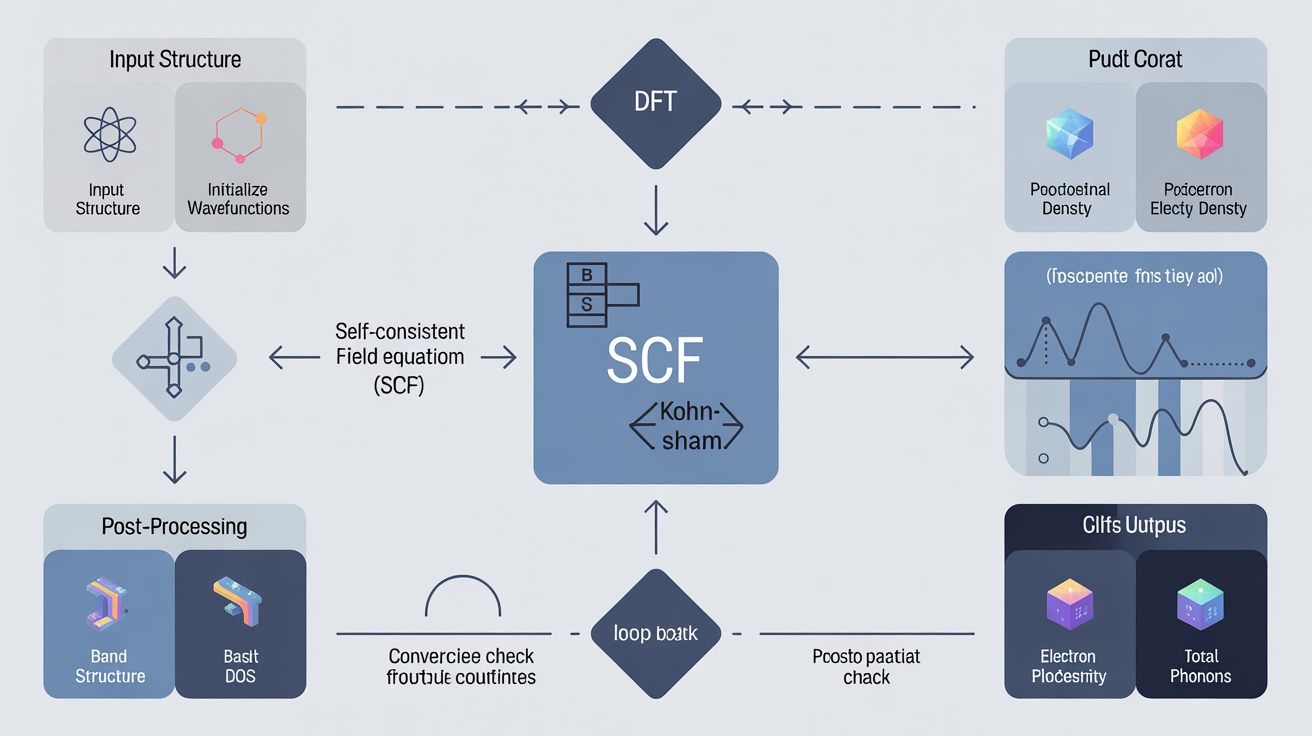
Quantum ESPRESSO's modular architecture consists of several integrated packages, with PWscf (Plane-Wave Self-Consistent Field) serving as the computational engine for ground-state calculations. The code excels at computing electronic band structures, density of states, phonon dispersion relations, and structural optimization for crystalline materials. Its plane-wave basis set approach, combined with pseudopotentials, allows efficient treatment of periodic systems ranging from simple metals to complex oxides and two-dimensional materials.
The software implements multiple exchange-correlation functionals, including local density approximation (LDA), generalized gradient approximation (GGA), and hybrid functionals. For systems with strong electron correlation, it supports DFT+U corrections and can interface with many-body perturbation theory codes for more accurate excited-state properties.

Practical Workflow for Materials Property Prediction
A typical Quantum ESPRESSO workflow begins with structural relaxation to find the equilibrium atomic geometry. Users define the crystal structure in the input file, specifying lattice parameters, atomic positions, and k-point sampling density. The code then iteratively minimizes forces and stresses until convergence criteria are met. This relaxed structure serves as the foundation for subsequent property calculations.
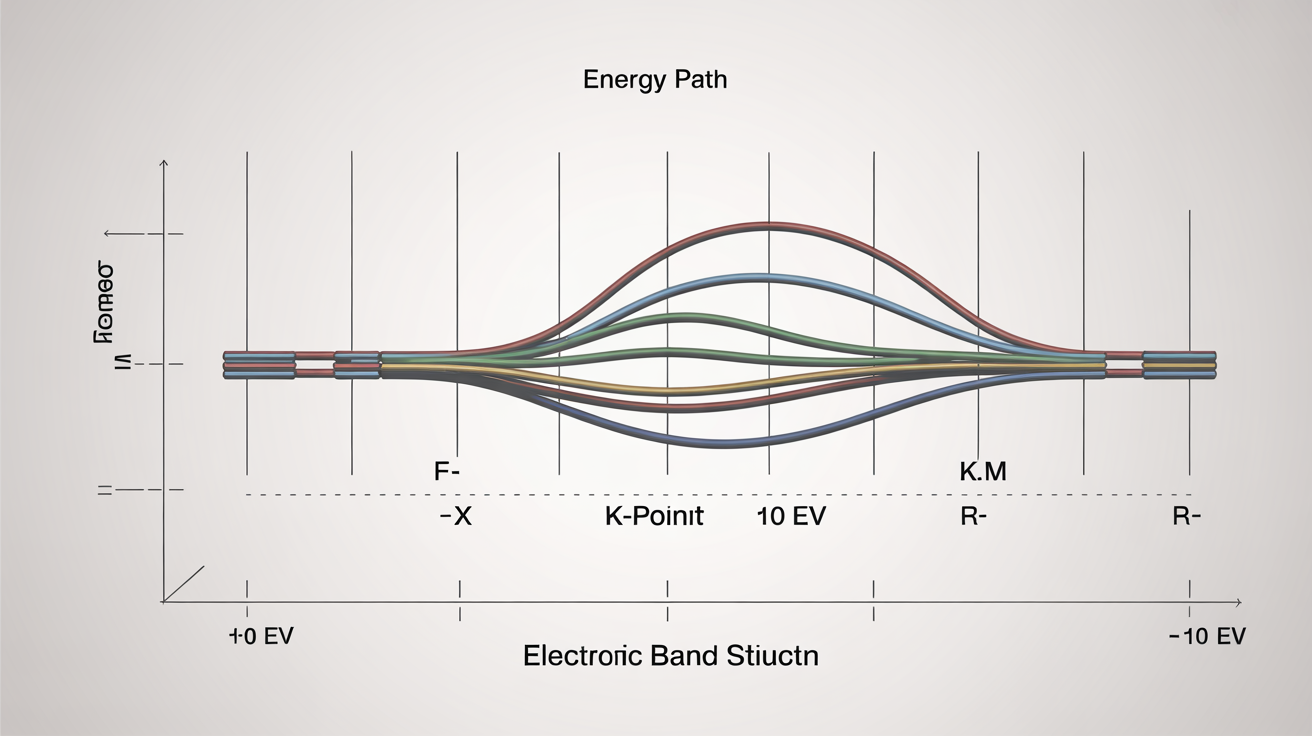
For electronic properties, practitioners commonly perform self-consistent field calculations followed by non-self-consistent runs along high-symmetry k-paths to generate band structures. The code's post-processing tools extract density of states projections, enabling identification of orbital contributions to electronic states near the Fermi level—critical information for understanding conductivity, optical properties, and chemical bonding.
Phonon calculations through density functional perturbation theory (DFPT) represent another strength of Quantum ESPRESSO. Rather than using finite-difference methods that require multiple supercell calculations, DFPT computes the dynamical matrix directly from linear response theory. This approach efficiently yields phonon dispersion curves, thermodynamic properties, and electron-phonon coupling constants essential for understanding superconductivity and thermal transport.
Performance Optimization and Scalability
Achieving computational efficiency with Quantum ESPRESSO requires careful attention to convergence parameters. The plane-wave energy cutoff must be systematically tested—typically ranging from 30 to 100 Ry depending on pseudopotential hardness. K-point mesh density similarly demands convergence testing, with denser grids needed for metallic systems and accurate density-of-states calculations.
The code demonstrates excellent parallel scalability on high-performance computing clusters through hybrid MPI/OpenMP parallelization. For large systems, users can exploit parallelization over k-points, plane-wave components, and linear algebra operations. Modern GPU acceleration support further enhances performance for computationally demanding calculations.
Integration with Materials Discovery Workflows
Quantum ESPRESSO integrates seamlessly with high-throughput computational materials science frameworks. The AiiDA (Automated Interactive Infrastructure and Database for Computational Science) platform provides workflow management and provenance tracking for Quantum ESPRESSO calculations, enabling systematic exploration of chemical spaces. Similarly, the Materials Project and other databases extensively use Quantum ESPRESSO for generating reference data on material stability, electronic structure, and elastic properties.
The code's open-source nature and extensive documentation have fostered a vibrant user community. Regular workshops and tutorials help newcomers navigate the learning curve, while active development continues to expand capabilities—recent additions include support for non-collinear magnetism, spin-orbit coupling, and advanced functionals for van der Waals interactions.
Practical Considerations and Best Practices
Successful Quantum ESPRESSO simulations require understanding the trade-offs between accuracy and computational cost. Pseudopotential selection significantly impacts both—ultrasoft and projector-augmented wave (PAW) pseudopotentials reduce plane-wave cutoff requirements but may sacrifice transferability. The SG15 ONCV and PseudoDojo libraries provide well-validated pseudopotential sets for most elements.
For complex materials with localized d or f electrons, standard DFT often fails to predict correct electronic structure. In such cases, DFT+U corrections or hybrid functionals become necessary, though at substantially increased computational expense. Benchmarking against experimental data or higher-level theory helps validate the chosen methodology.
Conclusion
Quantum ESPRESSO represents a mature, versatile platform for first-principles materials simulation. Its combination of computational efficiency, methodological rigor, and extensive functionality makes it indispensable for modern materials research. As computational resources continue to expand and algorithmic improvements emerge, Quantum ESPRESSO will remain central to predictive materials design and discovery efforts.
For researchers seeking to implement first-principles calculations in their workflow, Quantum ESPRESSO offers an accessible entry point with professional-grade capabilities. The extensive documentation at quantum-espresso.org and active user forums provide essential support for both novice and expert users.