
VASP for Materials Property Prediction: Accurate Electronic Structure Calculations

The Vienna Ab initio Simulation Package (VASP) has established itself as the gold standard for first-principles electronic structure calculations in materials science. Unlike general-purpose molecular dynamics codes, VASP specializes in solving the quantum mechanical equations that govern electron behavior in materials, enabling researchers to predict material properties from fundamental physics without empirical parameters.
Core Capabilities and Methodology
VASP implements density functional theory (DFT) using plane-wave basis sets and pseudopotentials, a combination that offers exceptional accuracy for periodic crystalline systems. The software employs the projector augmented-wave (PAW) method, which provides an optimal balance between computational efficiency and precision. This approach allows VASP to handle systems ranging from simple metals to complex oxides and two-dimensional materials with consistent reliability.
The code's iterative self-consistent field (SCF) solver efficiently converges electronic ground states, typically requiring 20-50 iterations for well-behaved systems. VASP's sophisticated charge density mixing algorithms, including Kerker and Broyden methods, accelerate convergence even for challenging cases like strongly correlated materials or systems with narrow band gaps.

Predicting Mechanical and Electronic Properties
One of VASP's most powerful features is its ability to calculate elastic constants and mechanical properties through stress-strain relationships. By applying small lattice deformations and computing the resulting stress tensors, researchers can extract the full elastic tensor, bulk modulus, shear modulus, and Young's modulus. This capability proves invaluable for screening candidate materials for structural applications or understanding mechanical failure mechanisms at the atomic level.
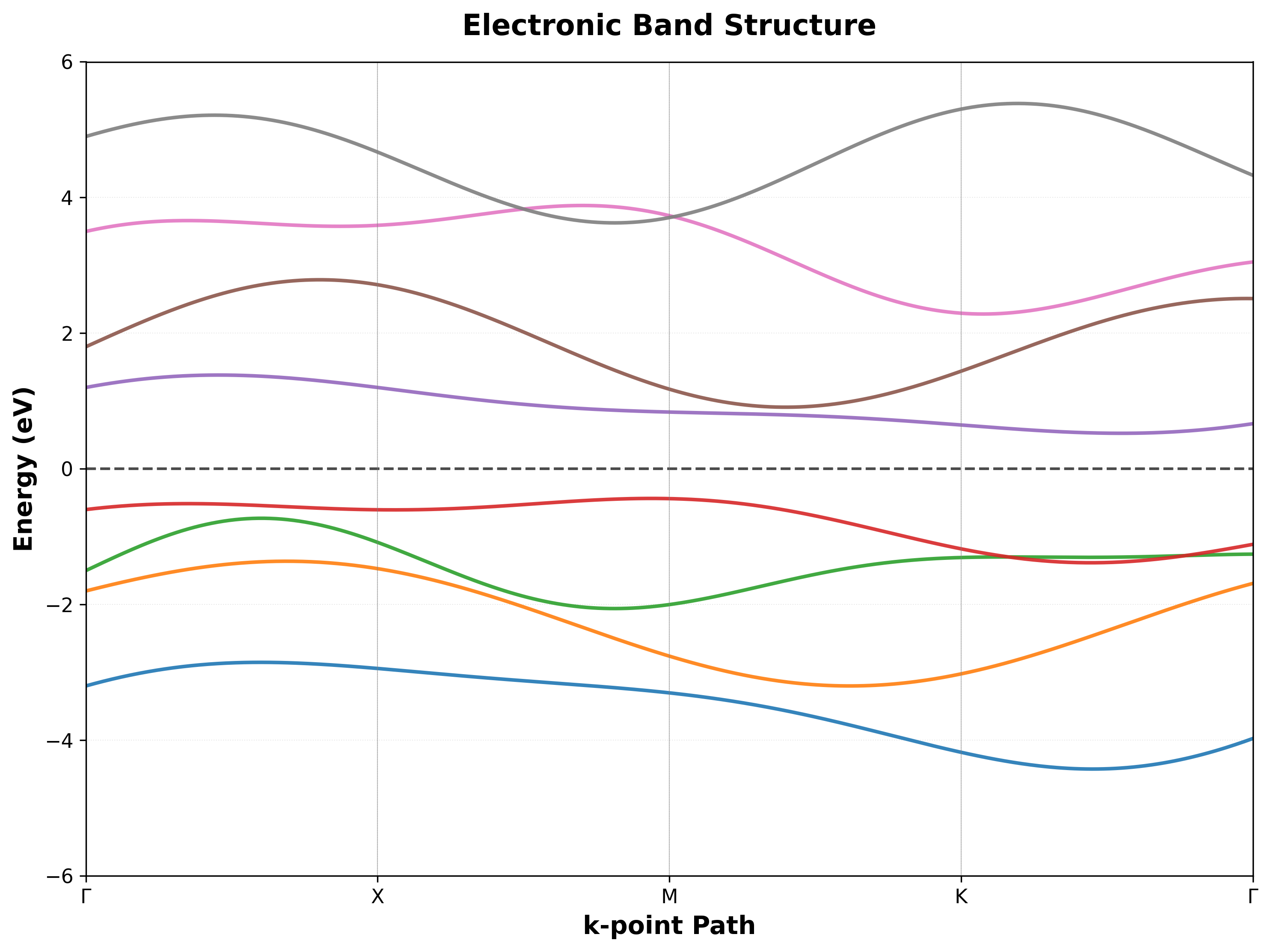
For electronic properties, VASP excels at computing band structures, density of states, and optical absorption spectra. The code supports hybrid functionals like HSE06, which correct the well-known band gap underestimation problem of standard DFT. This makes VASP particularly suitable for semiconductor research, where accurate band gaps are critical for predicting device performance.
Advanced Features for Materials Discovery
VASP's implementation of the nudged elastic band (NEB) method enables calculation of reaction pathways and energy barriers for atomic diffusion, chemical reactions, and phase transformations. This functionality is essential for understanding kinetic processes in battery materials, catalysts, and solid-state reactions. The climbing-image NEB variant provides precise saddle-point geometries without requiring additional calculations.
The software also supports ab initio molecular dynamics (AIMD), combining quantum mechanical accuracy with finite-temperature sampling. AIMD simulations reveal temperature-dependent phenomena like thermal expansion, phase stability, and ionic conductivity that static calculations cannot capture. For battery electrolytes and superionic conductors, AIMD provides insights into ion transport mechanisms that guide experimental synthesis efforts.
Computational Considerations and Best Practices
Achieving reliable results with VASP requires careful convergence testing. Users must systematically verify that total energies converge with respect to plane-wave cutoff energy (typically 400-600 eV for PAW potentials) and k-point mesh density. The Monkhorst-Pack scheme generates well-distributed k-point grids, with denser meshes needed for metals than insulators due to Fermi surface integration requirements.
Memory and computational demands scale with system size and basis set completeness. A typical calculation for a 100-atom supercell with 500 eV cutoff requires 16-32 CPU cores and 64-128 GB RAM, completing in several hours to days depending on convergence difficulty. High-performance computing clusters with fast interconnects are essential for production calculations on complex materials.
Integration with Materials Databases and Workflows
VASP integrates seamlessly with high-throughput computational frameworks like the Materials Project and AFLOW, which have generated millions of DFT calculations to accelerate materials discovery. Python interfaces such as pymatgen and ASE (Atomic Simulation Environment) enable automated workflow construction, from structure generation through post-processing and visualization. These tools allow researchers to screen thousands of candidate materials for specific property targets, dramatically accelerating the materials design cycle.
The combination of VASP's accuracy, comprehensive feature set, and ecosystem of supporting tools makes it indispensable for modern computational materials science. Whether predicting novel battery cathodes, designing catalysts for sustainable chemistry, or understanding the electronic properties of quantum materials, VASP provides the quantitative foundation for materials innovation.
Further Resources
- Official VASP documentation: https://www.vasp.at/wiki/
- Materials Project database: https://materialsproject.org/
- pymatgen materials analysis library: https://pymatgen.org/
- ASE atomic simulation environment: https://wiki.fysik.dtu.dk/ase/